Lecture 1: A deterministic model for epidemics¶
Introduction¶
In this topic we are going to be thinking about modelling the spread of infectious disease in some population. We will do this using a classic compartment model, where we divide the population into three types:
- Susceptible - uninfected individuals with no prior immunity,
- Infected - infected and infectious individuals,
- Recovered - immune to reinfection.
More detailed model structures exist, but we will focus on this initial simple model for now. We can then think about how individuals move between these three compartments to describe the dynamics of the model. Again, we will start with the simplest possible (yet biologically reasonable) structure where the only two processes are infection (Susceptible -> Infected) and recovery (Infected -> Recovered).
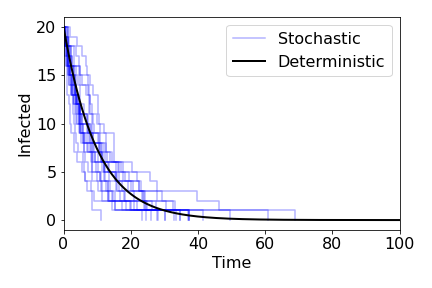
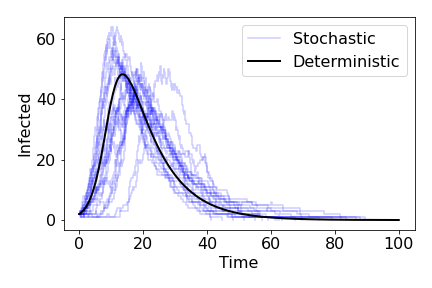
This is known as the SIR model and has formed the cornerstone of modelling infectious diseases in plant, animal and human populations since the 1920s. In general, the model is built as a set of ordinary differential equations. This is a deterministic model, where we assume the population sizes are large and that time and the variables are continuous. An important feature of this type of model is that, if we take the same parameter values and initial conditions, we will get the exact same time-course every time we run the model, and there is no randomness involved. Of course, the real world is messier than this, and we would expect a degree of inherent randomness - the precise time an infection takes place, for example. An alternative approach that captures this inherent randomness is a stochastic model. Now the same parameters and initial conditions will lead to different time-courses every time. Stochastic models can also give us more accuracy over what happens when population numbers are small and assume that events cause discrete changes in the numbers of individuals (rather than continuous changes to the density). However, their inherent randomness often makes them harder to analyse.
The deterministic model¶
Although our focus is to be on the stochastic implementation of a model of disease spread, it is useful for us first to get a good understanding of its deterministic counterpart. As such, let us assume that the population is large, and that both time and the population densities are continuous. We can then describe the dynamics of the model using the following ordinary differential equations,
\begin{align} &\frac{dS}{dt} = -\beta SI\label{si1}\\ &\frac{dI}{dt} = \beta SI - \gamma I\label{si2}\\ &\frac{dR}{dt} = \gamma I \end{align}with the total population, $N=S+I+R$. Note that $dN/dt = dS/dt+dI/dt+dR/dt=0$, and so the total population stays at a constant size. This is a reasonable assumption for studying a short-term epidemic outbreak in long-lived populations. Because of this we can eliminate one variable, with $R=N-(S+I)$ and ignore the $dR/dt$ equation. We should also stress that while we will often think of these variables as numbers, they are actually densities (since they are continuous variables).
While this is a fairly simple looking system (two variables and two parameters), there is no explicit solution. However, we can explore the qualitative behaviour of the model by identifying equilibria of the system and their stability. Moreover, we can use numerical routines to simulate the possible dynamics, as we will do in the first computer lab.
Considering these equations, we can see that the only equilibrium is when $I=0$ and $S$ may take any value $<N$. We therefore seem to have a continuum of non-isolated equilibria. To assess the stability here we need to take the Jacobian of the system - a matrix made up of partial derivatives of our ODEs:
\begin{align*} J=&\left( \begin{array}{cc} \frac{\partial (dS/dt)}{\partial S} &\frac{\partial (dS/dt)}{\partial I} \\ \frac{\partial (dI/dt)}{\partial S} & \frac{\partial (dI/dt)}{\partial I} \end{array} \right)\\ =&\left( \begin{array}{cc} -\beta I & -\beta S \\ \beta I & \beta S-\gamma \end{array} \right)\\ \end{align*}Since $I=0$ the bottom-left entry is $0$, meaning we can read the eigenvalues off as the entries on the main diagonal. We therefore have $\lambda_1=0$ and $\lambda_2=\beta S-\gamma$. The fact that one eigenvalue is 0 confirms our assessment that we have a line of non-isolated equilibria. The stability of each point now just depends on $\lambda_2$. We can see that for $S>\gamma/\beta$, the eigenvalue is positive and that point is unstable, while for $S<\gamma/\beta$, the eigenvalue is negative and that point is stable. Overall then, we expect the dynamics to eventually reach an equilibrium where $I=0$ and $S$ is some value less than $\gamma/\beta$ (but the precise value depends on the initial conditions).
This implies that in the long-run, we would expect the population to reach a point with no infected hosts i.e. the system will become disease-free. In the long-run this is often the case with emerging diseases - they will eventually burn out - but it is still important for us to know whether there can be an epidemic, when an initially small amount of infection causes a large outbreak of disease in the population (even if it eventually tends to zero).
For an epidemic to occur, we need to have $dI/dt>0$ initially. Before an outbreak, the initial densities are $S(0)\approx N,I(0)\approx0$. This gives,
\begin{equation} \frac{dI}{dt}=(\beta N-\gamma)I \end{equation}- $\beta N-\gamma<0$ $\implies$ disease dies out.
- $\beta N-\gamma>0$ $\implies$ epidemic.
Note that $\beta N-\gamma>0 \implies \beta N/\gamma>1$. This fraction is an important measure of how quickly a disease spreads at early points in the epidemic, and is known as the basic reproduction ratio, or $R_0$. Provided $R_0>1$, we see a classic epidemic curve, of initially exponential growth, slowing to a peak and then gradually tailing off towards 0.
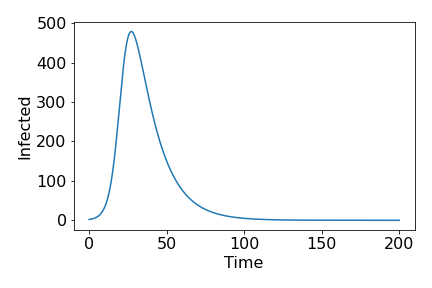
Figure 1. Example of epidemic curve from deterministic model. Paramters: $N=1000, \gamma=1/14, R_0=5$.
An extended model¶
We have introduced the simplest possible model for the spread of an infetcious disease, with just three possible compartments ($S$, $I$ and $R$) and only two mechanisms (infection and recovery). We have therefore built a lot of assumptions into the model, some of which are more obvious than others. Let us think about one particular extension, which is to allow immunity to wane at rate $\omega$, such that individuals who were in the recovered compartment can now return to being susceptible once again. We can then update our model as follows,
\begin{align} &\frac{dS}{dt} = -\beta SI+\omega R\\ &\frac{dI}{dt} = \beta SI - \gamma I\\ &\frac{dR}{dt} = \gamma I - \omega R. \end{align}Notice that we still have a constant population size, meaning we can still study only the first two equations, setting $R=N-S-I$. We now have,
\begin{align} &\frac{dS}{dt} = -\beta SI+\omega (N-S-I)\\ &\frac{dI}{dt} = \beta SI - \gamma I \end{align}We now find we have two possible equilibria. Firstly we can have $(S,I)=(N,0)$, the disease-free case. This time, however, the susceptible density takes a fixed value of $N$, because eventually everyone who was recovered will lose their immunity and return to being susceptible. The second equilibrium occurs at: $$(S,I)=\left(\frac{\gamma}{\beta},\frac{\omega\left(N-\frac{\gamma}{\beta}\right)}{\gamma+\omega}\right) $$ which after some re-arranging can be written as, $$(S,I)=\left(\frac{\gamma}{\beta},\frac{\omega\gamma}{\beta(\gamma+\omega)}(R_0-1)\right). $$
Clearly, then, the second equilibrium only exists for feasible values if $R_0>1$. It is left as a challenge for you to prove that the stability of each point depends on whether $R_0$ is larger than or less than 1.